Gas Adsorption
In this experiment you will learn about gas adsorption to a solid surface.
Theoretical background
Introduction
Gas adsorption has been studied theoretically for most of this century, and the simplest of the resulting theories provide the insight needed for most applications. We will investigate two such treatments, one attributed to Langmuir and one to Brunauer, Emmett and Teller (BET) and will apply their equations to our experimental data. We will investigate the adsorption of N2 at cryogenic temperatures onto common high-area supports such as charcoal and aluminum oxide. We will use this information to test simple adsorption theory, determine the specific area of the absorbent, and estimate the heat of adsorption of N2.
When a gas or vapor is brought into contact with a solid, part of it is taken up by the solid. This phenomenon is called adsorption. The phenomenon of adsorption was discovered over two centuries ago: The uptake of gases by charcoal was studied by C. W. Scheele in 1773 and by F. Fontana in 1777. In 1785, charcoal was found to decolorize' solutions by a surface adsorption mechanism.
When a molecule approaches a surface, it encounters a net attractive potential that is similar to the potential between two molecules and arises for the same reasons. However, a gas molecule near a surface is attracted not by a single molecule but by several closely spaced surface atoms. The adsorption of a molecule onto a solid surface is almost always an exothermic process. If we represent the gas molecule (also called the adsorbate), by A and the adsorption site on the surface by S, the process of adsorption can be represented as a chemical reaction:
The surface of an atomic solid has about 1015 atoms per cm2 of surface. If we assume that one molecule adsorbs on each atom of the solid surface, then there are 1015 sites on which molecules can adsorb. When one molecule is adsorbed on each site, the surface is said to be covered with a monolayer.
The solid that takes up the gas is called the adsorbent, and the gas or vapor taken up on the surface is called the adsorbate. It is not always easy to tell whether the gas is inside the solid or merely at the surface because most practical adsorbents are very porous bodies with large internal surfaces. It is not possible to determine the surface areas of such materials by optical or electron microscopy because of the size and complexity of the pores and channels of the material. The gas adsorption itself, however, can be used to determine the accessible surface area of most absorbents.
Gas adsorption is of practical consequence to engineers and chemists in many ways. It can provide a convenient, cheap and reusable method for fluid purification. Gas masks used from World War I to the present day are an example of the utility of charcoal as an adsorbent. More significantly, perhaps, the phenomenon of surface adsorption has been used to modify the rates of product yields of chemical reactions through heterogeneous catalysis. For a catalyst to be useful, it must have a large surface area, bind the reactants quickly and effectively, stabilize the activated complex, and release the products of the reaction. Thus the attractive potentials of surface for various molecules, as well as the total surface area of the catalyst are extremely important properties of potential catalytic materials.
The extent of the covered surface is expressed as the fractional coverage, θ:
where n is the number of adsorbed molecules (mol), nS is the number of surface sites (i.e., the number of molecules required to create a monolayer) (mol).
Physisorption and Chemisorptions
Physical adsorption, or physisorption, involves a van der Waals interaction between the adsorbate and the substrate. A particle is adsorbed because it lowers its potential energy upon forming an intermolecular bond. Since the change in adsorption enthalpy is very small (in the range of 20 kJ/mol), chemical bonds remain intact and the adsorbed molecule retains its identity.
In chemical adsorption, or chemisorption, a molecule sticks to the surface by forming a chemical (usually covalent) bond- and tends to find a site that maximizes its coordination number with the substrate. The enthalpy in the case of chemisorption is much higher than for physiosorption (in the range of 200 kJ/mol). A chemisorbed molecule may be torn apart due to unsatisfied valencies of the surface atoms. This is one reason why solid surfaces catalyze reactions.
Almost all adsorption processes (chemisorption and physisorption) are exothermic: The translational freedom of the adsorbed molecule decreases relative to the molecule free in solution, so that ΔS < 0. In a spontaneous process ΔG = ΔH − TΔS < 0, so that ΔH must be negative. The process is endothermic only when the translational energy gained by breaking the chemical bond is greater than that lost due to the adsorption to the surface.
The enthalpy of adsorption is a convenient way to distinguish physisorption from chemisorptions. The enthalpy depends upon the extent of surface coverage, θ. If the particles repel each other, a large surface coverage tends to increase ΔH (make it less negative), and vice versa.
Adsorption Isotherms
The free gas and the adsorbed gas are in dynamic equilibrium, and the fractional coverage of the surface depends on the pressure, P, of the overlying gas. The variation of θ with pressure at a chosen temperature is called the adsorption isotherm.
The Langmuir Isotherm
The Langmuir isotherm is based on three assumptions:
- Adsorption cannot proceed beyond monolayer coverage.
- All surface sites are equivalent and can accommodate, at most, one adsorbed atom.
- The ability of a molecule to adsorb at a given site is independent of the occupation of neighboring sites and there are no interactions between adsorbed molecules.
The dynamic equilibrium is:
The rate constants are ka for adsorption and kd for desorption. At equilibrium, the rate of change of θ due to adsorption should be equal to the rate of change of θ due to desorption, so there is no net change of fractional coverage θ. The dependence of θ on pressure is called the Langmuir isotherm, and it is described by the following equation:
where p is the partial pressure of the gas. In this experiment we will use only nitrogen gas so that p=P. K is the ratio between the adsorption and desorption constants (Torr-1). By measuring P and n the number of surface site and the adsorption rate can be calculated:
In an ideal gases, a plot of P/n versus P is a linear function with a slope of 1/nS and intercept at 1/KnS.
When the adsorption is accompanied by dissociation, the Langmuir isotherm becomes more weakly dependent on pressure:
The shape of the Langmuir isotherm is shown in Figure 1. As can be seen, θ increases with pressure, until gas is forced on to every available site of the surface and θ → 1.
 (b)
(b)
The isosteric enthalpy of adsorption, ΔH°ads, is the standard enthalpy of adsorption at a fixed surface coverage (constant θ). The adsorption rate K is dependent on temperature and is an equilibrium constant; therefore, using the van't Hoff equation we can determine ΔH°ads:
Equation 4 can be written also as:
For a constant θ, at equilibrium, the following equation applies:
Thus, equation 7 can be written as:
After some rearrangement, we get:
Therefore, a plot of lnP vs. 1/T should be a straight line of slope ΔH°ads.
The BET Isotherm
In the Langmuir model, it was assumed that adsorption only occurs on unoccupied substrate adsorption sites. We will now remove this restriction. If the initial adsorbed layer can act as a substrate for further adsorption, instead of the isotherm leveling off to a saturated value at high pressures, it can rise indefinitely. The most widely used isotherm dealing with multilayer adsorption was derived by Stephen Brunauer, Paul Emmet and Edward Teller, and is called the BET isotherm.
In the BET model, the enthalpy change in forming the second (and higher) layers are assumed to be identical to the enthalpy of condensation of the gas. Molecules in the first layer have a different energy owing to the direct interaction with the surface. The fractional coverage, θ, under these assumptions becomes:
In this expression P* is the saturation pressure of the gas (i.e., the vapor pressure of the liquid at that temperature), and c is a constant:
The constant c depends on both ΔH°des and ΔH°vap, the enthalpies of desorption from the monolayer and of vaporization of the liquid adsorbate, respectively. c is large when the enthalpy of desorption from a monolayer is large compared with the enthalpy of vaporization of the liquid adsorbate.
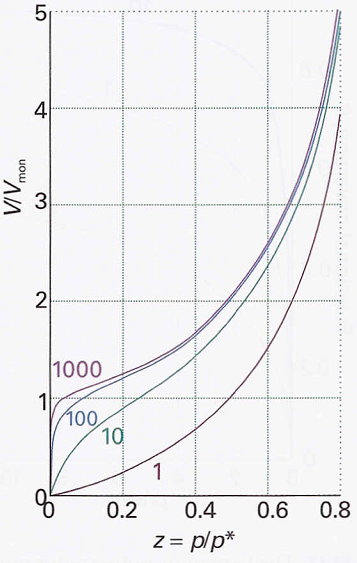
The shapes of the BET isotherms for different values of c are shown in Figure 2. The BET isotherm rises indefinitely as the pressure is increased because there is no limit to the amount of material that may condense when multilayer coverage can occur. A BET isotherm is not accurate at all pressures, since it underestimates the extent of adsorption at low pressures and overestimates it at high pressures. However, it is still widely used in industry to determine the surface areas of solids.
Equation 12 may be rearranged to:
If z/[n(1-z)] is plotted against z, then c and ns can be determined using the slope and intercept of the plot.
Adsorption Surface Area
The surface area covered by an adsorbed monolayer can be easily calculated. The surface area is:
where NA is Avogadro's number and σ is the surface area of a single particle. For Nitrogen σ = 0.16 nm2.
Adsorption and Desorption Rates
The rate at which a surface is covered by adsorbate depends on the ability of the substrate to absorb the thermal energy of the incoming particle as it crashes on to the surface. If the energy is not dissipated quickly, the particle migrates over the surface until a vibration expels it into the overlying gas or it reaches an edge. The sticking probability, s, is defined as the proportion of collisions with the surface that successfully lead to adsorption:
A small value of s indicates that many collisions are needed before a molecule sticks successfully to the surface. The sticking probability is related to the fractional surface coverage:
where s0 is the sticking probability on a perfectly clean surface.