| Ph.D.: |
Hebrew
University of Jerusalem, 1976 |
| Phone: |
(Office):
+972-3-6408984
(Lab): +972-3-6406695
(Home): +972-3-6094174
Fax (Office): +972-3-6406834
|
| E-mail: |
shoshbn@tauex.tau.ac.il
|
| Room#: |
Room
613 (office); room 611 (lab) |
|
|
|
The Discovery of p97/Cdc48 and its Role in ERAD
|
|
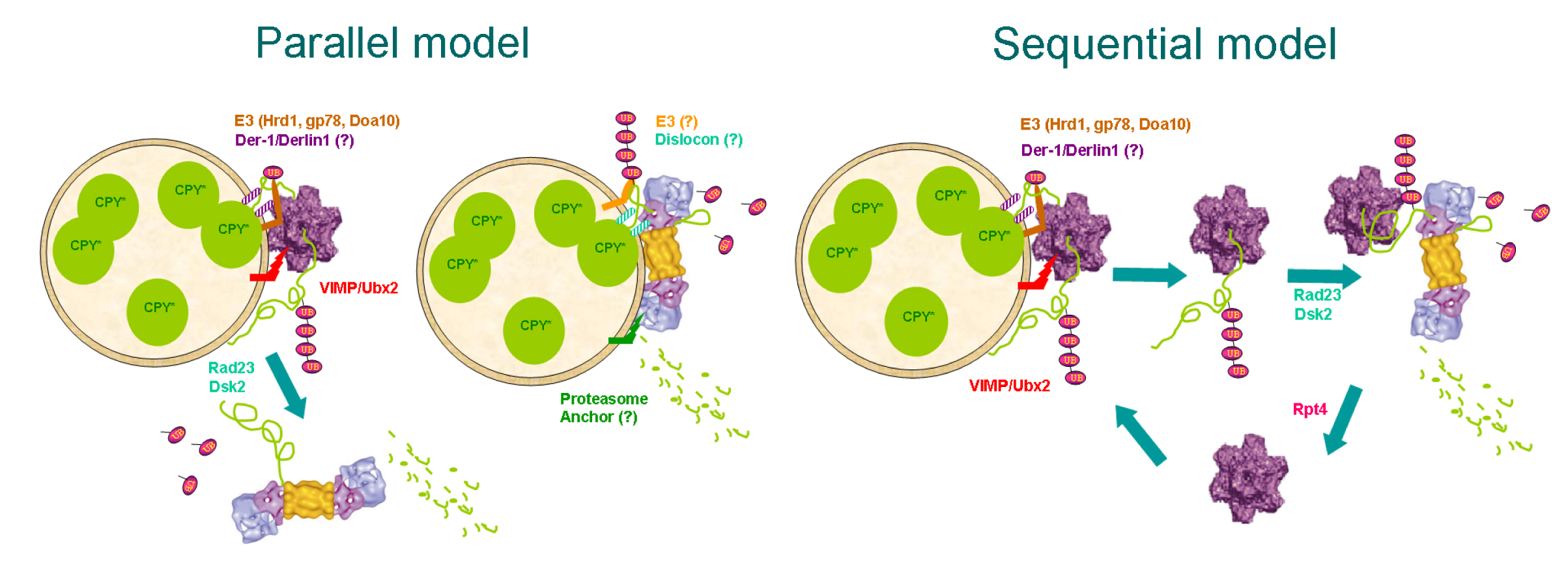
In 2000 we
discovered that the p97, and its conserved
yeast homologue Cdc48, is an essential
component of ERAD that takes part in the
elimination of virtually all ERAD
substrates (Rabinovich et al., 2002). The
p97/Cdc48 is a cytosolic homo-hexameric
AAA-ATPase that functions as a molecular
machine.
Initially, we found p97 in
association with a luminal ERAD substrate
and subsequently demonstrated the role of
its homologue Cdc48 in the degradation in
yeast of two well-established ERAD
substrates, membrane 6myc-Hmg2 and the
luminal CPY* (Rabinovich et al. 2002). To
date, p97/Cdc48 is a hallmark of ERAD,
although it also participates in several
other cellular functions (Bar-Nun, 2005).
Being an ATPase that resides in the
cytosol, p97/Cdc48 provides the driving
force for dislocating ERAD substrates from
the ER back to the cytosol, as the
stabilized luminal ERAD substrate CPY*
remained trapped within the ER lumen in
the temperature-sensitive cdc48-10
mutant
(Elkabetz et al., 2004).
Interrelations
between Cdc48 and proteasomal AAA-ATPases:
The 26S proteasome complex is composed of a 20S proteolytic chamber
and a 19S regulatory cap. Several
AAA-ATPases (known in yeast as Rpt1-6)
form a hetero-hexameric ring at the base
of this 19S regulatory
particle. We have shown that the
proteolytic activity of the proteasome is
dispensable for the dislocation a luminal
ERAD substrate (Elkabetz et al., 2004) but
two of the 19S regulatory particle
subunits, Rpt2 and Rpt4, are essential for
ERAD. While Rpt2 is required for
degradation of every proteasomal
substrate, since it gates the entry of
substrates into the proteolytic chamber,
Rpt4 is essential for the elimination only
of ERAD substrates and is dispensable for
degradation of cytosolic proteins (Lipson
et al., 2008). Interestingly, Rpt4 is
involved in dislocation of ERAD
substrates, a role already assigned to
Cdc48 (Elkabetz et al., 2004).
Further experiments on the
interrelations between these two
AAA-ATPases suggest that Cdc48 extracts
the substrate from the ER, while Rpt4 is
required for transferring the substrate
from Cdc48 to the proteasome (Lipson et
al., 2008). We also studied the gating of
the 20S catalytic particle and showed that
degradation of cytosolic and ERAD
substrates was similarly accelerated upon
truncation of the N-termini of the 20S
subunits
a3 and
a7 known to gate
the proteolytic chamber (Rabinovich et
al., 2006).
Cdc48 Suppressors: Ssz1 and the link between ERAD
and PDR:
In its role in ERAD, Cdc48
collaborates with Ufd1 and Npl4, forming a
Cdc48-Ufd1-Npl4 complex. In genetic screens for
suppressors of cdc48 temperature-sensitive
mutants, we have identified SSZ1 and show
that it upregulates Cdc48 via the pleiotropic drug
resistance (PDR) network. A pSSZ1 plasmid
restored the impaired ERAD of the membrane
substrates 6myc-Hmg2 in
yeast cells carrying mutations in
cdc48,
ufd1
and
npl4,
while deletion of the SSZ1 gene had no
effect. Ssz1p activates Pdr1p, the PDR master
regulator. Indeed, plasmids of PDR1 or its
target gene RPN4 increased the levels of
mutant Cdc48 protein and restored ERAD in the
cdc48-10
temperature-sensitive mutant. Rpn4 regulates
transcription of proteasome subunits but also of
CDC48, thus RPN4 deletion
abolished ERAD. However, the diminished proteasome
level in
Drpn4
was sufficient for degrading a cytosolic
substrate, whereas the impaired
ERAD-M was the result of
diminished levels of Cdc48 and indeed, ERAD was
restored by expression of pCDC48. The
restored ERAD-M in the hypomorphic strains
of the Cdc48 partners ufd1-2 and npl4-1
by the pCDC48 plasmid, and in cdc48-10
temperature-sensitive mutant by the pcdc48-10
plasmid, combined with the finding that neither pSSZ1
nor pcdc48-10 restored ERAD-L of CPY*-HA,
support our conclusion that Ssz1 suppressing
effects is brought about by upregulating
Cdc48(Bosis et al., 2009). These findings uncover
a regulatory link between PDR, which induces
membrane transporters for efflux of cytotoxic
compounds, and ERAD, which eliminates damaged
proteins generated by such compounds, and extend
our knowledge on the coordination of cellular
networks that are responsible for coping with
stress.
 |
Enter here specific template content
|




